IX.5 There is a great deal of selectively neutral genetic polymorphism at the molecular level in natural populations
At the level of the DNA sequence, the degree of polymorphism in the population can be quantified in several ways.It is frequently characterized by the fraction of polymorphic genes, i.e. the content of genes that occur in at least two alleles in the studied population.As, if we had a sufficiently large sample of the studied individuals, we would probably find that practically all genes are polymorphic in this sense, only those genes whose commonest allele occurs, for example, in a maximum of 99% of the individuals in the population, are generally considered to be polymorphic. The heterozygosity index (H), which is basically the frequency of heterozygotes in the population, is another commonly employed measure of the degree of polymorphism in the population.For the individual genes, this index is usually calculated from the frequencies of the individual alleles:
H = 1 - Σ xi2,
where xi is the frequency of the i-th allele in the population.Thus, a population containing a large number of alleles with the same frequency has the largest H value.The average heterozygosity index for the given population can be calculated on the basis of the heterozygosity indices as the arithmetic mean for the individual genes.If the heterozygosity index is calculated on the basis of sequence data, it is also sometimes called thegene diversity index.The nucleotide (aminoacid) diversity index (B) can also be calculated on the basis of sequence data; this corresponds to the average number of nucleotide (aminoacid) differences between all the pairs of alleles in the sample divided by the length of the sequences of the relevant alleles.The average number of pair differences Π can be calculated for the whole population or, to be more precise, for the population sample, as
Π = [ 1/n(n-1)/2 ]Σi<jΠij,
where n is the number of observed sequences (so that n (n -1)/2 is the number of various pairs of sequences) and Πij is the number of differences between the i-th and j-th sequence.
As was mentioned at the beginning of the chapter, the results obtained by the methods of molecular biology indicate that the individual members of a particular species differ in the occurrence of various nucleotides in a great many positions on their genes and thus the gene pool of all species of organisms contains an enormous amount of genetic polymorphism.A considerable part of this polymorphism in the gene pool of a species and the gene pools of the individual populations apparently exists because it is selectively neutral and selectively neutral traits can persist in the population for a very long time.Most selectively neutral traits are eventually eliminated from the population by genetic drift or genetic draft; however, mutation processes generate new polymorphisms by that time.Mutations in all parts of the genome that do not code any protein, especially in pseudogenes, i.e. inactivated genes, and thus nonfunctional copies of genes, are usually considered to be selectively neutral.The category of neutral mutations apparently also includes a large part of mutations in introns and also part of synonymous mutations, i.e. nucleotide substitutions, for example in the third positions of a large percentage of nucleotide triplets.Because of the degeneracy of the genetic code, i.e. that fact that, in a great many cases, 2-6 various triplets mostly differing in the nucleotide in the third position code the same aminoacid, the substitution of a nucleotide here does not lead to substitution of an aminoacid in the coded protein.However, in absolute values, even these categories of mutations need not and apparently do not have zero selection coefficients (Akashi 1995).In the case of introns, a change in the primary sequence can lead to a change in the efficiency of their splicing at the RNA level or changes in the secondary structures of the introns can also directly affect the intensity and rate of transcription of the relevant genes.For synonymous mutations, a change in the synonymous codon can affect the rate of the transcription, as the individual tRNA serving for translation of the synonymous codons are not present in the cell in the same number of copies and translation of mRNA occurring with participation of rarer tRNA is necessarily slower (see also XI.6.2.5).The fact that the primary structure of the gene affects not only the primary structure of the resulting proteins but, through the effect on the secondary structure of the DNA and mRNA, also the processes of regulation of DNA transcription and break-down of the relevant RNA, may be important.Thus, synonymous mutations do not affect the aminoacid sequence of the relevant protein but can substantially affect its concentration in the cell.Studies performed on drosophila did actually show that unevenness in the use of the individual synonymous codons is negatively correlated with divergence in synonymous mutations between related drosophila
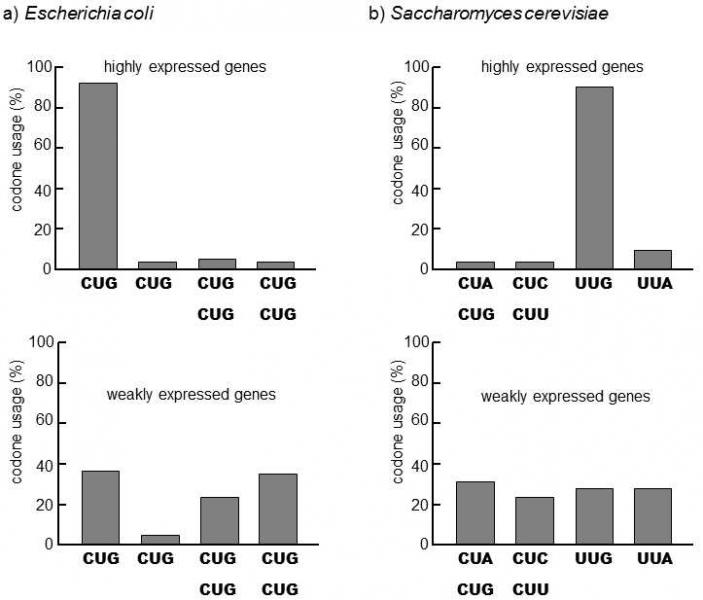
Fig. IX.14. Uneven use of the individual synonymous codons for leucine in strongly and weakly expressed genes. It is apparent from the graph that the use of the individual synonymous codons is far less even in strongly expressed genes than in weakly expressed genes. The most probable explanation is that it doesn’t much matter in poorly expressed genes if codons, whose relevant tRNA is present in a small number of copies in the cells, are used for coding leucine. The occurrence of these codons retards the translation of the particular mRNA, which could be detrimental in the case of strongly expressed genes. The efficiency of the selection is very high for the bacteria E. coli and the yeast S. cerevisiae, i.e. in species forming large populations. As a consequence, even synonymous mutations slightly reducing the rate of translations can be a subject of natural selection. Data according to Ikemura (1981, 1982), modified according to Li and Graur (2001).
species (Fig. IX.14)(Sharp & Li 1989). As the degree of unevenness in the use of synonymous codons is also positively correlated with the intensity of expression of the individual genes, it is apparent that stronger negative selection acts against synonymous mutations in more intensely expressed genes compared to genes that are not expressed strongly (Fig. IX.15) (Akashi 1994).
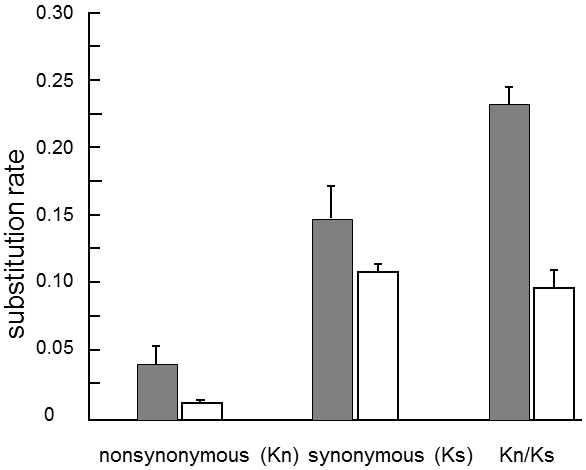
Fig. IX.15. Accelerated evolution of sequences of genes related to sexual reproduction. The column graph depicts the differences between genes whose function is related (dark columns) or not related (white columns) to sexual reproduction, in the substitution rates in nonsynonymous positions(Kn), in synonymous positions (Ks) and in the ratio of the substitution rates in nonsynonymous and synonymous positions (Kn/Ks). Pairs of genes derived from two species of drosophila (D. melanogaster and D. simulans), were compared, specifically 10 genes related to sexual reproduction and 46 other genes. The substitution rates are expressed in the numbers of substitutions per nonsynonymous or synonymous site from the time of branching off of the two species. The error bars depict the standard error of the mean. According to Singh and Kulathinal (2000).
Draft translation from: Evoluční biologie, 2. vydání (Evolutionary biology, 2nd edition), J. Flegr, Academia Prague 2009.
The translation was not done by biologist, therefore any suggestion concerning proper scientific terminology and language usage are highly welcomed. You can send your comments to flegr cesnet [dot] cz. Thank you.
cesnet [dot] cz. Thank you.